
Welcome to the Kay Lab
Our laboratory has long been interested in the development of gene transfer vectors for gene therapy as well as manipulating non-coding RNAs for therapeutic purposes. A major interest has been in unraveling the mechanism of viral vector transduction in vivo. Our work has focused on two vector systems, mini-circles and recombinant AAVs (rAAV). Using gene transfer vectors, we studied the potential of using transcriptional-based RNAi to treat human disease. Our work uncovered key mechanistic insights into how non-coding miRNAs are loaded into active RISC complexes in mammals.
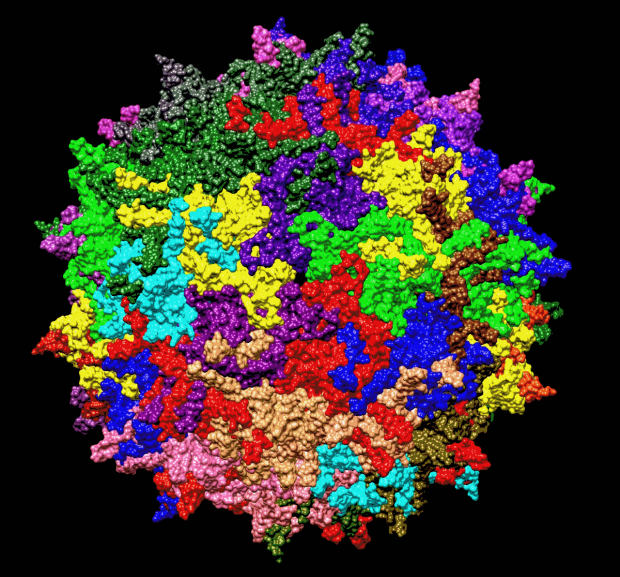
AAV capsid currently used in many gene therapy trials
These parameters are crucial for evaluating how effective particular AAV capsids will be before they reach the clinic and help resolve current mysteries around AAV capsid efficacy.

The expanding world of small non-coding RNAs derived from tRNAs
We discovered new classes of small RNAs derived from tRNA and non-coding RNAs that may be generated by RNA-directed RNA transcription in vertebrates. This is one of several types of non-coding RNA we are investigating.